
Medical device manufacturers face various pre and post market challenges. Some of the challenges are prominent, in spite of, establishing design controls in the development stages, it is difficult to foresee all the probable challenges unless we use the global standards, Design of Experiments (DoE), methodological processes are part of our plan.
Here are precise, FDA oriented, and non-generic classification of the major problems
Design related Challenges
Challenges left before the company entails ambiguity of design inputs, poor traceability, weak risk integration, and uncontrolled design changes, which can lead to FDA observations, and recalls
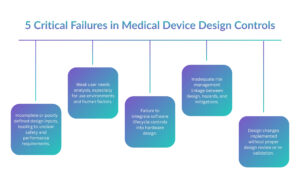
Common Design Failures and Root Causes
People often encounter major problems in the area of design because of untraceable design inputs, risk analysis that is either delayed or superficial, overreliance on predicate devices, and even human factors that are neglected, all of which usually result in post-market issues and recalls.
How Design Issues Lead to FDA Recalls
One of the main reasons for medical devices FDA recalls are the incorrect outputs coming from the devices in actual use, wrong alarm or display interpretations, and mechanical failures caused by not properly validated design tolerances, thus emphasizing the need for complete design validation and risk management.
Example 1: Infusion Pump
One of the major design problems was the inaccuracy of the flow rate at certain pressure conditions, which was brought about by variable clinical environments in the design inputs and ultimately led to a Class II recall because of the risk of either under or over infusion. Infusion systems and other critical care devices were among those facing recalls or urgent safety notices, pointing out that underlying gaps in design robustness and simulation of use conditions remain key causes of post-market failures.
Example 2: Wearable Cardiac Monitor
When the patient moved, signal dropout occurred due to the insufficient design validation for the dynamic conditions, which caused Fan DA recall and enforced design remediation.
Example 3: Diagnostic Software Device (SaMD)
Algorithm bias affecting specific patient populations, due to poorly defined clinical performance requirements, triggered an FDA safety communication and post-market corrective actions.
Example 4: Particularly, recalls occurred in continuous glucose monitors when receivers did not alert patients during dangerous glucose swings, leading to patient injury. The root causes included weak risk analyses and inadequate real-world testing that failed to detect alarm failures before clearance.
Taken together, these trends are consistent with recall data indicating software malfunction and incomplete design controls as common FDA intervention triggers.
FDA Verification, Validation, and Software Testing Challenges
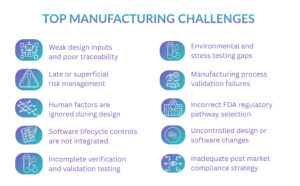
In the device development process, one can expect such common issues as non-final verification, system validation, software testing, and documentation to be hardly being up to the mark. These problems might result in the malfunction of devices, their withdrawal from the market, and strict actions being taken by the FDA.
Important product Testing Challenges for Medical Devices and Software
- Verification testing that does not fully cover design inputs
- Validation testing performed on non-representative samples
- Incomplete software validation documentation
- Cybersecurity testing is treated as optional
- Failure to validate manufacturing processes impacting device performance
How Improper Testing Causes Device Failures and Recalls?
When design assumptions are unverified, software defects appear in clinical use, environmental stress failures occur post-market, or manufacturing variability causes inconsistent performance, FDA recalls often cite a “failure to adequately validate the device design under actual or simulated use conditions.”
1: Surgical Robotic System
Software lag during complex procedures, caused by incomplete system-level validation only unit level testing, resulted in an FDA recall and mandatory software update.
2: Glucose Monitoring Device
Inaccurate readings at temperature extremes, due to insufficient environmental testing, led to a Class II recall and updated label restrictions.
3: Mobile Medical App
An app crash during data transmission, caused by insufficient stress and interoperability testing, resulted in an FDA warning letter and corrective action plan.
FDA Regulatory Pathway and Compliance Failures
What Happens When Certification Is Missing or Incorrect?
If a company does not get the right FDA certification, it can suffer from product seizure, import detention, warning letters, mandatory recalls, civil penalties, and losing access to the U.S. market. Typical
Issues related to enforcement are wrong predicate selection, selling devices beyond their cleared indications, and making software changes without new submissions.
Example 1: AI-Based Diagnostic Tool
The FDA took enforcement action, and finally the product was withdrawn because the marketing of an algorithm update without FDA review, which was a result of misunderstanding the impact of the software change, led to that situation.
Example 2: Class II Implantable Device
Because it failed in the 510(k) clearance process, the product was not introduced in the market, and efforts have been made beyond metamorphosis in order to come to substantial realization costed time and money to the manufacturer
Example 3: Connected Medical Device
The decision made by the Refuse-to-Accept (RTA) led to the submission being denied because the cybersecurity documentation was not included defying FDA demands, resubmissions, and delay the product to market
Final Takeaway
Based on the patterns observed in the FDA enforcement actions, the major factors responsible for product recalls and market failures are design flaws, inadequate testing, and mistakes in product certification, rather than the amount of documentation submitted. The technical development that includes design controls, thorough testing, and selection of the right FDA route will be the one to reap the benefits while the rest will suffer from delays, product recalls, and legal actions.
Bench Testing is where most programs lose time and money. Bench testing often fails in EMC/EMI/others because it does not reflect real-world use conditions or worst-case scenarios expected by the FDA. Software and SaMD products struggle even more due to incomplete verification strategies, poor cybersecurity justification, and shallow clinical evaluation logic. Validation testing is frequently misaligned with intended use claims, leading to FDA questions that cannot be answered without additional studies. The core issue is not regulation, it is improper or late alignment. Teams that treat FDA expectations as an afterthought end up testing to satisfy auditors instead of proving the device actually works safely and consistently. The only reliable way through is to build regulatory thinking into design and test planning from the start, not after the prototype development.






